Figures titles links
Implication of senescence in giant cell arteritis
The pathogenic role of cellular senescence in the context of Giant Cell Artiritis (GCA), an age-related, autoinflammatory and autoimmune syndrome has not been addressed before. This study identified a high proportion of senescent cells (mainly fibroblasts and macrophages) with an inflammatory phenotype in GCA arteries. The presence of senescent cells was associated with the tissue inflammatory bulk via IL-6-dependent pathways, suggesting a potential implication of senescence in disease pathogenesis. These results set the ground for the use of senolytic treatments as a possible complementary strategy in the clinical management of GCA.
Machine learning algorithms to predict drug responses
A. Schematic representation of the study design and bioinformatics pipeline. (a) Dataset: the full data set was constructed using the GDSC and CCLP databases (see also Supplemental Fig. 1 in ref 1). (b) Model construction: Association Rule Mining (ARM) was used to generate testable hypotheses of genes associated with sensitivity or resistance to specific drugs (left panel). (c) Validation: our models were validated computationally and in a variety of in vitro experimental settings.
B. A machine learning workflow was designed to predict drug response and survival of cancer patients. All pipelines were trained on a large panel of cancer cell lines and tested in clinical cohorts. Deep Neural Networks outperformed other machine learning algorithms and captured pathways that link gene expression with drug response (2).
1) Vougas et al. Machine learning and data mining frameworks for predicting drug response in cancer: An overview and a novel in silico screening process based on association rule mining. Pharmacol Ther 2019, 203: 107395.
2) Sakellaropoulos et al. A deep learning framework for predicting response to therapy in cancer. Cell Rep 2019, 29(11): 3367-3373.e4.
A fluorophore-conjugated reagent enabling rapid detection, isolation and live tracking of senescent cells
Cellular senescence is a stress-response mechanism implicated in various physiological processes, diseases, and aging. Current detection approaches have partially addressed the issue of senescent cell identification in clinical specimens. Effective methodologies enabling precise isolation or live tracking of senescent cells are still lacking. In-depth analysis of truly senescent cells is, therefore, an extremely challenging task. Here we report (1) the synthesis and validation of a fluorophore-conjugated, Sudan Black-B analog (GLF16), suitable for in vivo and in vitro analysis of senescence by fluorescencemicroscopy and flow cytometry and (2) the development and application of a GLF16-carrying micelle vector facilitating GLF16 uptake by living senescent cells in vivo and in vitro. The compound and the applied methodology render isolation of senescent cells an easy, rapid, and precise process. Straightforward nanocarrier-mediated GLF16 delivery in live senescent cells comprises a unique tool for characterization of senescence at an unprecedented depth.
Cellular Senescence: Defining a Path Forward
The Hallmarks of the Senescence Phenotype. Senescent cells exhibit the following four interdependent hallmarks: cell-cycle withdrawal, macromolecular damage, secretory phenotype (SASP), and deregulated metabolism (1).
1) Gorgoulis et al. Cellular Senescence: Defining a Path Forward. Cell 2019, 179(4):813-827.
Cellular senescence and cardiovascular diseases
Cellular senescence has been the subject of focused research for over two decades, however it was not until recently that the role of senescence in cardiovascular disease (CVD) started to be explored. A body of evidence has shown that cellular senescence constitutes a stress-mediated pathophysiological mechanism implicated in a wide range of CVDs at the cellular and molecular level. In this review, we recapitulate key molecular and clinical findings supporting the involvement of senescence in heart disease and discuss current clinical strategies aimed at eradicating the detrimental effects of senescence on cardiac homeostasis. Based on recent evidence, we additionally address how the advent of the senotherapeutics field, in conjunction with the development of novel senescence detection tools in tissues and biological fluids may now facilitate effectively combating CVDs.
Integrating the DNA damage and protein stress responses during cancer development and treatment
A model depicting how oncogene induced replication stress aids the progressive formation of certain hallmarks of cancer (early events: steps 1-5), while paving the way for, angiogenesis, evasion from immune surveillance, invasion and metastasis (late events-6). Specifically, oncogenic activation acts as a force that pushes the cell away from its equilibrium point. Activated oncogenes lead to replication stress either directly by deregulating the replication machinery or indirectly via affecting metabolic pathways (1, 2). DNA lesions resulting from oncogene activation stimulate the DNA damage response pathway to promote repair and impose the tumorigenic barriers of apoptosis and senescence. In the event of a perturbed DNA damage response, cells accumulate genomic instability, proteotoxic and mitotic stress (3, 4). Failure to elicit apoptosis or escape from senescence (5) can lead to oncogenic transformation and primary tumor formation. These early events can also pave the way for later events including angiogenesis, evasion from immune surveillance, invasion and metastasis. This is a link that requires further investigation (see text for details). Strike, hourglass: over time, genomic instability shapes the stages for cancer progression. ?: a potential link that requires further investigation [see text in: ref 1 for details and additional references].
1] Gorgoulis et al. Integrating the DNA damage and protein stress responses during cancer development and treatment. J Pathol 2018, 246(1): 12-40.
Machine learning: a tool to shape the future of medicine
The constant evolution of biomedicine, biophysics and biochemistry has enabled scientists to investigate and study each cell identity via analyzing the transciptome and its kinetics, the chromatin accessibility patterns but also via investigating the structure of proteins and RNA. Taking all this under consideration, scientists have developed algorithms and machine learning (ML) schemes that take advantage of the current state-of-the-art approaches to predict the cell states, discover the exact 3D structure of proteins and RNA and most importantly evaluate personalized medicine approaches via predicting drugs and specific immunotherapy treatments. Moreover, the recent advances in ML and chemo-informatics have also paved the way for drug repurposing models, thus evaluating and establishing in silico novel treatments. The aim of this chapter is to provide and analyze the mathematics behind such ML techniques and review the current applications being developed that walk side by the side with the continuous progress of biosciences.
The non cell-autonomous role of mutant p53 gain-of-function: reprogramming the microenvironment
Colon cancers cells harboring mutations in TP53 release exosomes that contain miR1246 which in turn are received by surrounding macrophages. The latter alter their phenotypic features, leading to secretion of anti-inflammatory cytokines along with factors implicated in epithelial and mesenchymal transition (EMT), which fuel tumorigenesis in this setting (1, 2).
1) Cooks et al. When mutant p53 fires up. Cancer Cell & Microenvironment 2014, 1: e135.
2) Cooks et al. Mutant p53 cancers reprogram macrophages to tumor supporting macrophages via exosomal miR-1246. Nat Commun 2018, 9(1): 771.
Pulmonary infection by SARS-CoV-2 induces senescence accompanied by an inflammatory phenotype in severe COVID-19: possible implications for viral mutagenesis
In patients with severe COVID-19, alveolar type II (AT2) cells that are infected with SARS-CoV-2 undergo senescence accompanied also by an inflammatory phenotype. In vitro recapitulation of SARS-CoV-2 infection in two-dimensional (2D) cellular systems and in a three-dimensional alveosphere system derived from alveolar type 2 (AT2) cells, also induced senescence and inflammation. Importantly, infected cells can act as a source of mutagenesis for SARS-CoV-2, since they exhibit increased levels of the APOBEC deaminating enzymes, which can mediate viral mutations. Overall these findings support that SARS-CoV-2 induced senescence may be an important driver of COVID-19 severity, disease persistence and the emergence of new viral strains. Finally, SARS-CoV-2-induced senescence may justify the implementation of senotherapeutics for the treatment of COVID-19 patients and possibly for long-COVID syndrome.
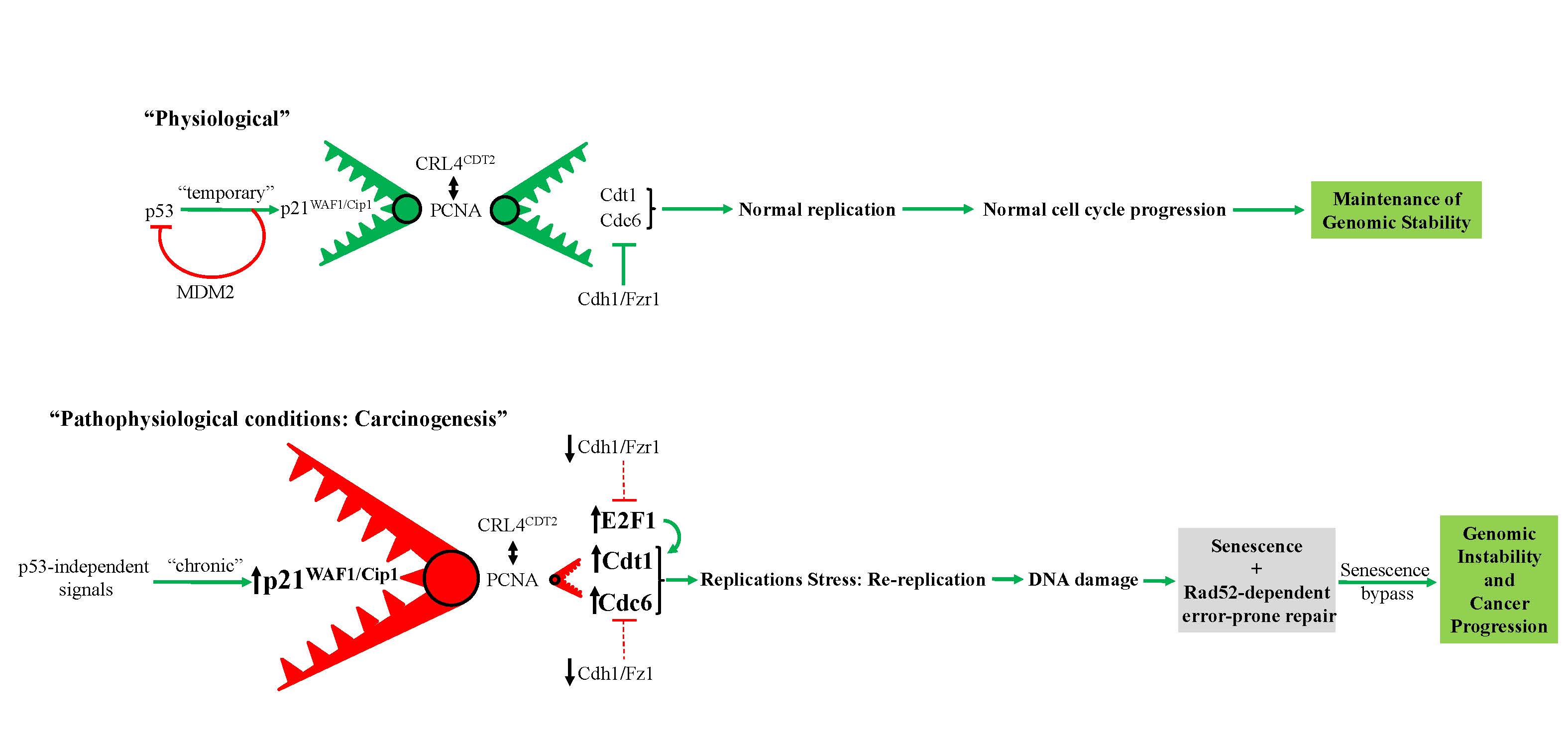
Prolonged expression of p21WAF1/Cip1 in p53-null cells as a driving force for cancer progression
Proposed model depicting prolonged p53-independent p21WAF1/CIP1 oncogenic action. The model shows how p53-independent p21WAF1/CIP1 expression fuels Rad52 dependent error-prone DNA double strand break repair promoting genomic instability (1, 2). Particularly, sustained p21WAF1/CIP1 induction in a p53-independent manner destabilizes the genome through two autonomous but complimentary routes: i) on one hand p21WAF1/CIP1 suppresses its degradation module, CRL4-CDT2, possibly by oversaturating it, as p21WAF1/CIP1 has the strongest PCNA-binding affinity, leaving their other targets, including CDT1, CDC6 and E2F1, free to perform their functions (see below Figure). By upregulating the replication licensing factors CDT1 and CDC6, the cells expressing p21WAF1/CIP1 acquire the capacity to re-replicate, generate DSBs, eventually driving a genome-destabilizing process. (Dashed lines depict ineffective pathway in below Figure), ii) on the hand p21WAF1/CIP1 leads to increased levels of nucleotide lesions mediated by elevated reactive oxygen species (ROS) (2). Given the negative impact exerted by p21WAF1/CIP1 on the error free nucleotide repair mechanisms (BER and NER), a significant proportion of such base lesions escape unrepaired. This creates an additional repair “load” to the error prone repair mechanism of TLS, which is further compromised by p21WAF1/CIP1 overexpression, leading to a decreased SNS load and in favor of DSBs. In turn, this further increases the DSB burden generated also through the re-replication step (i) (1). As components of SDSA are down-regulated, a shift to Rad52-mediated error prone DNA repair takes place by invoking the BIR and SSA repair routes, fueling genomic instability. This repair switch is mediated by a shift in the balance between Rad51 and Rad52 levels as the former is suppressed by E2F4 and the latter is induced by E2F1 (1). Overall these events occur throughout a senescence-like phase during which the error-prone DNA repair process takes place, forming a genetic landscape that allows a subpopulation of p21WAF1/Cip1–expressing cells to escape senescence, generating clones with aggressive features and increased chemo-resistance that promote cancer development. DSB: DNA double strand break, BER: base excision repair, NER: nucleotide excision repair, TLS: translesion DNA synthesis and repair, SDSA: synthesis-dependent strand annealing, BIR: break-induced repair, SSA: single strand annealing.
1) Galanos et al. Chronic p53-independent p21 expression causes genomic instability by deregulating replication licensing. Nat Cell Biol 2016, 18(7): 777-89.
2) Galanos et al. Mutational signatures reveal the role of RAD52 in p53-independent p21-driven genomic instability. Genome Biol 2018, 19(1): 37.
RASSF1A-mediated mechanism controlling tumor dedifferentiation and aggressive oncogenic behavior
This study is a continuation of many previous research articles on lung cancer development that represented the first investigation target, from the initiation of our research group (MCG). Particularly, it shows that the RASSF1A tumor suppressor uncouples the NOTCH-HES1 axis by triggering SNURF/RNF4-mediated HES1 ubiquitination. Loss of RASSF1A promotes cancer stemness via NOTCH-independent HES1 stabilization and confers resistance to γ-secretase inhibitors (GSI). This is the first evidence of crosstalk between the Hippo and Notch pathways, through the RASSF1A tumor suppressor. The study provides insight into the clinical setting where the use of GSIs constitutes a productive therapeutic approach in oncology.
Oncogenic Cdc6 as a molecular switch during cancer development
Model describing the ability of oncogenic Cdc6 to act as a molecular switch. (a and b) Cdc6 acting as a molecular switch at the CDH1 (E-cadherin) (a) and INK4/ARF (b) locus (see Discussion in ref 1). Ori: replication origin.
1) Sideridou et al. Cdc6 expression represses E-cadherin transcription and activates adjacent replication origins. J Cell Biol 2011, 195(7): 1123-40.
Escape from oncogene-induced senescence
The manuscript provides evidence on how senescent cells escape from oncogene induced senescence, an important tumor suppressor mechanism, facilitating tumor progression. Particularly the authors demonstrate that a recurrent chromosomal inversion harboring the circadian gene BHLHE40 is sufficient to drive escape from oncogene-induced senescence. The inversion is the outcome of oncogene-mediated genomic instability followed by chromatin refolding changes that activate the gene, leading to cell cycle re-entry and aggressive behavior. These findings support that replication stress-induced genomic instability is the causative factor underlying ‘‘escape’’ from oncogene-induced senescence and that targeting senescent cells can be of major clinical importance by eliminating a potential source of recurrence.
The role of Cdc6 in cancer progression
CDC6 acts as a prototypical oncogenic stimulus operating in a bimodal manner (1-4): i) triggering replication stress, fueling genomic instability (1,2,3), ii) functioning as a transcriptional repressor by switching off the INK4A/ARF and CDH1 (E-cadherin) loci, disrupting cell cycle progression and cell-to-cell communication (3,4).
1) Karakaidos et al. Overexpression of the replication licensing regulators hCdt1 and hCdc6 characterizes a subset of non-small-cell lung carcinomas: synergistic effect with mutant p53 on tumor growth and chromosomal instability--evidence of E2F-1 transcriptional control over hCdt1. Am J Pathol 2004, 165(4): 1351-65.
2) Liontos et al. Deregulated overexpression of hCdt1 and hCdc6 promotes malignant behavior. Cancer Res 2007, 67(22): 10899-909.
3) Sideridou et al. Cdc6 expression represses E-cadherin transcription and activates adjacent replication origins. J Cell Biol 2011, 195(7): 1123-40.
4) Petrakis et al. Cdc6: a multi-functional molecular switch with critical role in carcinogenesis. Transcription 2012, 3(3): 124-9.
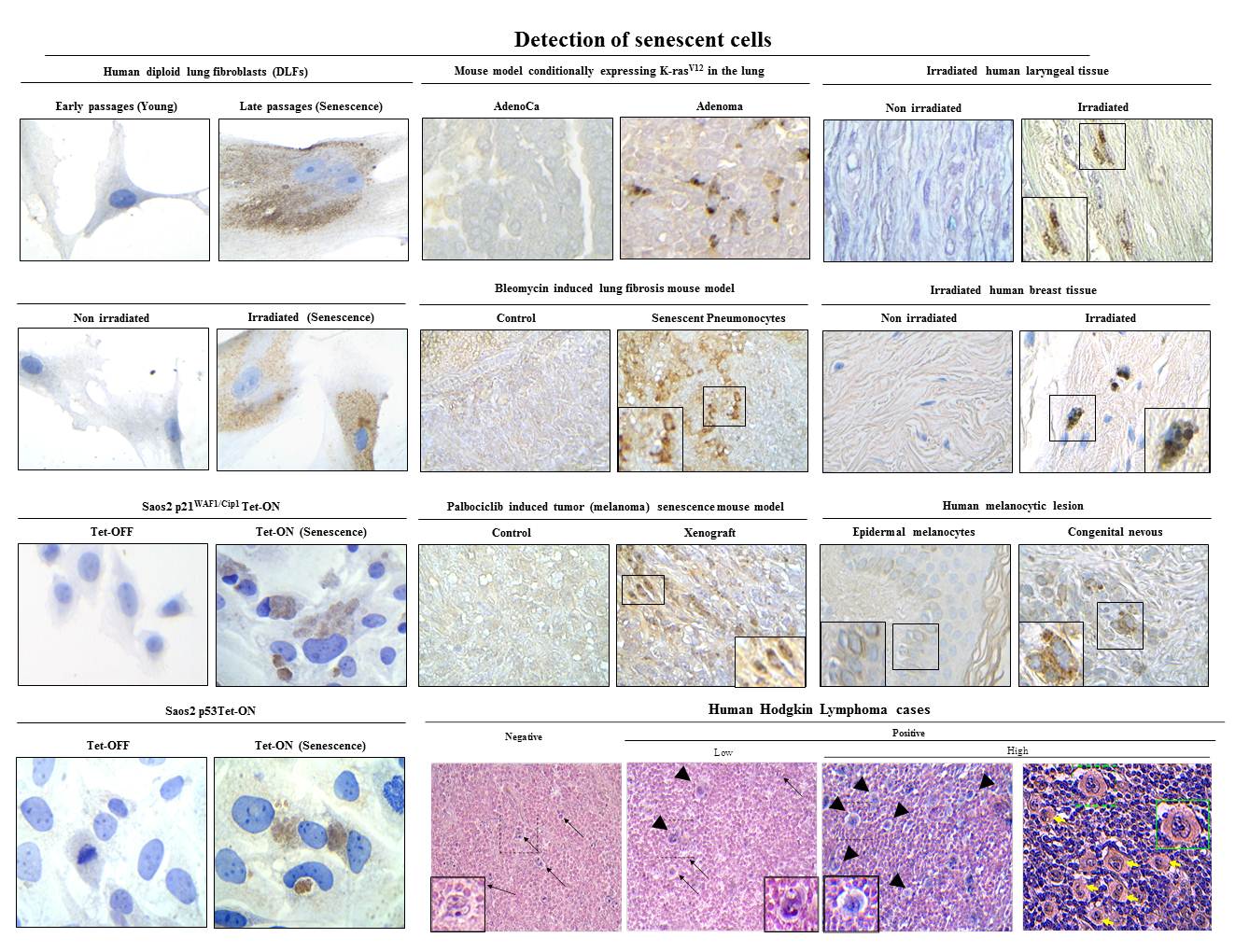
Algorithmic assessment of cellular senescence in experimental and clinical samples
Unequivocal identification and examination of cellular senescence remains highly difficult because of the lack of universal and specific markers. To overcome the limitation of measuring individual markers, we describe a detailed two-phase algorithmic assessment to quantify various senescence-associated parameters in the same specimen. In the first phase, we combine the measurement of lysosomal and proliferative features with the expression of general senescence-associated genes to validate the presence of senescent cells. In the second phase we measure the levels of pro-inflammatory markers for specification of the type of senescence. The protocol can help graduate-level basic scientists to improve the characterization of senescence-associated phenotypes and the identification of specific senescent subtypes. Moreover, it can serve as an important tool for the clinical validation of the role of senescent cells and the effectiveness of anti-senescence therapies.
Kohli J, Wang B, Brandenburg SM, Basisty N, Evangelou K, Varela-Eirin M, Campisi J, Schilling B, Gorgoulis V, Demaria M. Algorithmic assessment of cellular senescence in experimental and clinical specimens. Nature Protocols 2021. 2021 May;16(5):2471-2498.
An Oncogene-Induced DNA Damage Model for Cancer Development
Activated oncogenes disrupt normal proliferation, triggering replication stress, leading to DNA damage that stimulates the DNA damage response (DDR) pathway. Subsequently, DDR mobilizes the anti-tumor barriers of apoptosis and senescence.
As DNA damage accumulates, the cells’ capacity to repair in an error-free manner is overwhelmed, shifting repair to error-prone routes. The later fuels genomic instability, promoting cancer progression (1, 2, 3). Within this context, we have shown that DDR activation precedes ARF induction (4) and that ATM, a pivotal DDR upstream kinase, keeps in check ARF as a back-up anti-tumor response (5).
1) Gorgoulis et al. Activation of the DNA damage checkpoint and genomic instability in human precancerous lesions. Nature 2005, 434(7035): 907-13.
2) Bartkova et al. Oncogene-induced senescence is part of the tumorigenesis barrier imposed by DNA damage checkpoints. Nature 2006, 444(7119): 633-7.
3) Halazonetis et al. An oncogene-induced DNA damage model for cancer development. Science 2008, 319(5868): 1352-5.
4) Evangelou et al. The DNA damage checkpoint precedes activation of ARF in response to escalating oncogenic stress during tumorigenesis. Cell Death Differ 2013, 20(11):1485-97.
5) Velimezi et al. Functional interplay between the DNA-damage-response kinase ATM and ARF tumour suppressor protein in human cancer. Nat Cell Biol 2013, 15(8): 967-77.

Genomic instability — an evolving hallmark of cancer
Genomic instability as a hallmark of cancer. a) A proposed revision of the hallmarks of cancer to include genomic instability, and to consolidate the self-sufficiency in growth signals and insensitivity to anti-growth signals into the single hallmark of activated growth signalling. The secondary hallmarks (oxidative stress and proteotoxic stress) are shown separately. b) The temporal order by which the hallmarks are acquired in hereditary cancers. The establishment of genomic instability is probably the initiating event, which then facilitates the establishment of all the other hallmarks. c) The temporal order by which the hallmarks are acquired in sporadic (non-hereditary) cancers. Deregulation of growth-regulating genes can be the initiating event. This leads to DNA damage and DNA replication stress, which, in turn, lead to genomic instability and selective pressure for tumour suppressor p53 (TP53) inactivation. Loss of p53 function allows evasion from cell death, whereas the genomic instability provides a fertile ground for additional mutations that lead to the establishment of the remaining hallmarks, as in hereditary cancers (1). [Figure in part a) is modified, with permission, from REF 59 © (2009) Elsevier of (1)]
1) Negrini et al. Genomic instability--an evolving hallmark of cancer. Nat Rev Mol Cell Biol. 2010, 11(3): 220-8.
SenTraGor: a novel reagent to detect senescent cells
Design and synthesis of a novel chemical compound linked with biotin to detect senescent cells. (a) Overview of a pioneering method for senescent cell detection exploiting the specific reaction with lipofuscin of a novel chemical compound (GL13) linked with biotin. Beyond the histochemical capability of these compounds to stain senescent cells, the presence of biotin allows as a second-step application of an enhancing immunohistochemical-enzymatic detection reaction that provides increased sensitivity and recognition precision. (b) Structure of biotin and its particular moieties. (c) Synthesis of compound GL13 (commercially available as SenTraGor).
1) Evangelou et al. Robust, universal biomarker assay to detect senescent cells in biological specimens. Aging Cell 2017, 16(1): 192-197.
2) Myrianthopoulos et al. Senescence and senotherapeutics: a new field in cancer therapy. Pharmacol Ther 2019, 193: 31-49.

|
|
|
Prof. Vassilis G. Gorgoulis
Laboratory of Histology-Embryology
Chair of Clinical Molecular Pathology, Ninewells Hospital and School of Medicine
University of Dundee, Dundee, UK
Biomedical Research Foundation of the Academy of Athens
Faculty Institute for Cancer Sciences, University of Manchester, Manchester Centre for Cellular Metabolism,
EMBO member
European Academy
Academia Europaea member
Intelligencia.ai, 180 Varick Street, 6th Floor, New York, NY 10014, USA
Office Tel: 0030 210-7462352 |









News
Error: No articles to display


