Prolonged expression of p21WAF1/Cip1 in p53-null cells as a driving force for cancer progression
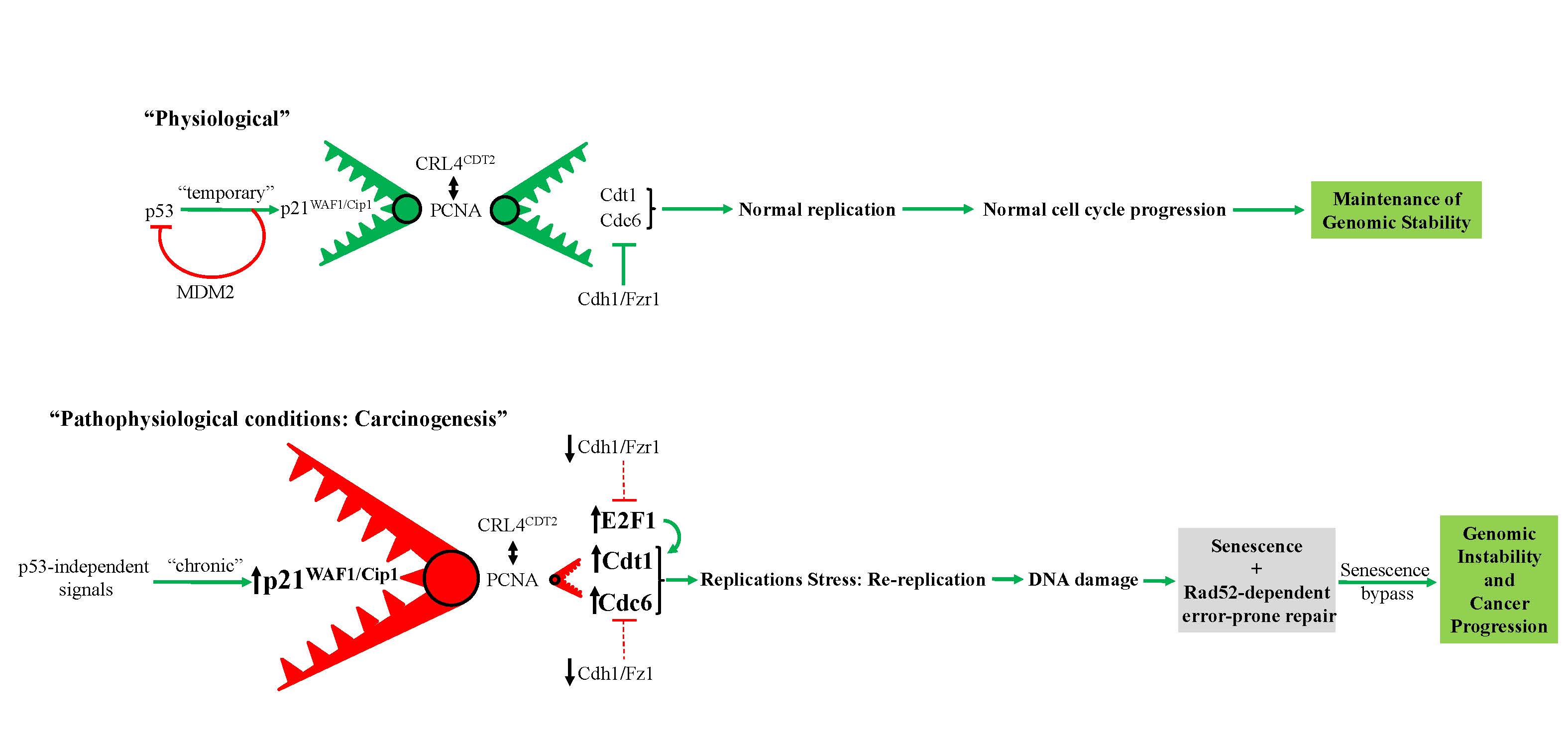
Proposed model depicting prolonged p53-independent p21WAF1/CIP1 oncogenic action. The model shows how p53-independent p21WAF1/CIP1 expression fuels Rad52 dependent error-prone DNA double strand break repair promoting genomic instability (1, 2). Particularly, sustained p21WAF1/CIP1 induction in a p53-independent manner destabilizes the genome through two autonomous but complimentary routes: i) on one hand p21WAF1/CIP1 suppresses its degradation module, CRL4-CDT2, possibly by oversaturating it, as p21WAF1/CIP1 has the strongest PCNA-binding affinity, leaving their other targets, including CDT1, CDC6 and E2F1, free to perform their functions (see below Figure). By upregulating the replication licensing factors CDT1 and CDC6, the cells expressing p21WAF1/CIP1 acquire the capacity to re-replicate, generate DSBs, eventually driving a genome-destabilizing process. (Dashed lines depict ineffective pathway in below Figure), ii) on the hand p21WAF1/CIP1 leads to increased levels of nucleotide lesions mediated by elevated reactive oxygen species (ROS) (2). Given the negative impact exerted by p21WAF1/CIP1 on the error free nucleotide repair mechanisms (BER and NER), a significant proportion of such base lesions escape unrepaired. This creates an additional repair “load” to the error prone repair mechanism of TLS, which is further compromised by p21WAF1/CIP1 overexpression, leading to a decreased SNS load and in favor of DSBs. In turn, this further increases the DSB burden generated also through the re-replication step (i) (1). As components of SDSA are down-regulated, a shift to Rad52-mediated error prone DNA repair takes place by invoking the BIR and SSA repair routes, fueling genomic instability. This repair switch is mediated by a shift in the balance between Rad51 and Rad52 levels as the former is suppressed by E2F4 and the latter is induced by E2F1 (1). Overall these events occur throughout a senescence-like phase during which the error-prone DNA repair process takes place, forming a genetic landscape that allows a subpopulation of p21WAF1/Cip1–expressing cells to escape senescence, generating clones with aggressive features and increased chemo-resistance that promote cancer development. DSB: DNA double strand break, BER: base excision repair, NER: nucleotide excision repair, TLS: translesion DNA synthesis and repair, SDSA: synthesis-dependent strand annealing, BIR: break-induced repair, SSA: single strand annealing.
1) Galanos et al. Chronic p53-independent p21 expression causes genomic instability by deregulating replication licensing. Nat Cell Biol 2016, 18(7): 777-89.
2) Galanos et al. Mutational signatures reveal the role of RAD52 in p53-independent p21-driven genomic instability. Genome Biol 2018, 19(1): 37.