
SenTraGor: a novel reagent to detect senescent cells
Design and synthesis of a novel chemical compound linked with biotin to detect senescent cells. (a) Overview of a pioneering method for senescent cell detection exploiting the specific reaction with lipofuscin of a novel chemical compound (GL13) linked with biotin. Beyond the histochemical capability of these compounds to stain senescent cells, the presence of biotin allows as a second-step application of an enhancing immunohistochemical-enzymatic detection reaction that provides increased sensitivity and recognition precision. (b) Structure of biotin and its particular moieties. (c) Synthesis of compound GL13 (commercially available as SenTraGor).
1) Evangelou et al. Robust, universal biomarker assay to detect senescent cells in biological specimens. Aging Cell 2017, 16(1): 192-197.
2) Myrianthopoulos et al. Senescence and senotherapeutics: a new field in cancer therapy. Pharmacol Ther 2019, 193: 31-49.

Machine learning algorithms to predict drug responses
A. Schematic representation of the study design and bioinformatics pipeline. (a) Dataset: the full data set was constructed using the GDSC and CCLP databases (see also Supplemental Fig. 1 in ref 1). (b) Model construction: Association Rule Mining (ARM) was used to generate testable hypotheses of genes associated with sensitivity or resistance to specific drugs (left panel). (c) Validation: our models were validated computationally and in a variety of in vitro experimental settings.
B. A machine learning workflow was designed to predict drug response and survival of cancer patients. All pipelines were trained on a large panel of cancer cell lines and tested in clinical cohorts. Deep Neural Networks outperformed other machine learning algorithms and captured pathways that link gene expression with drug response (2).
1) Vougas et al. Machine learning and data mining frameworks for predicting drug response in cancer: An overview and a novel in silico screening process based on association rule mining. Pharmacol Ther 2019, 203: 107395.
2) Sakellaropoulos et al. A deep learning framework for predicting response to therapy in cancer. Cell Rep 2019, 29(11): 3367-3373.e4.
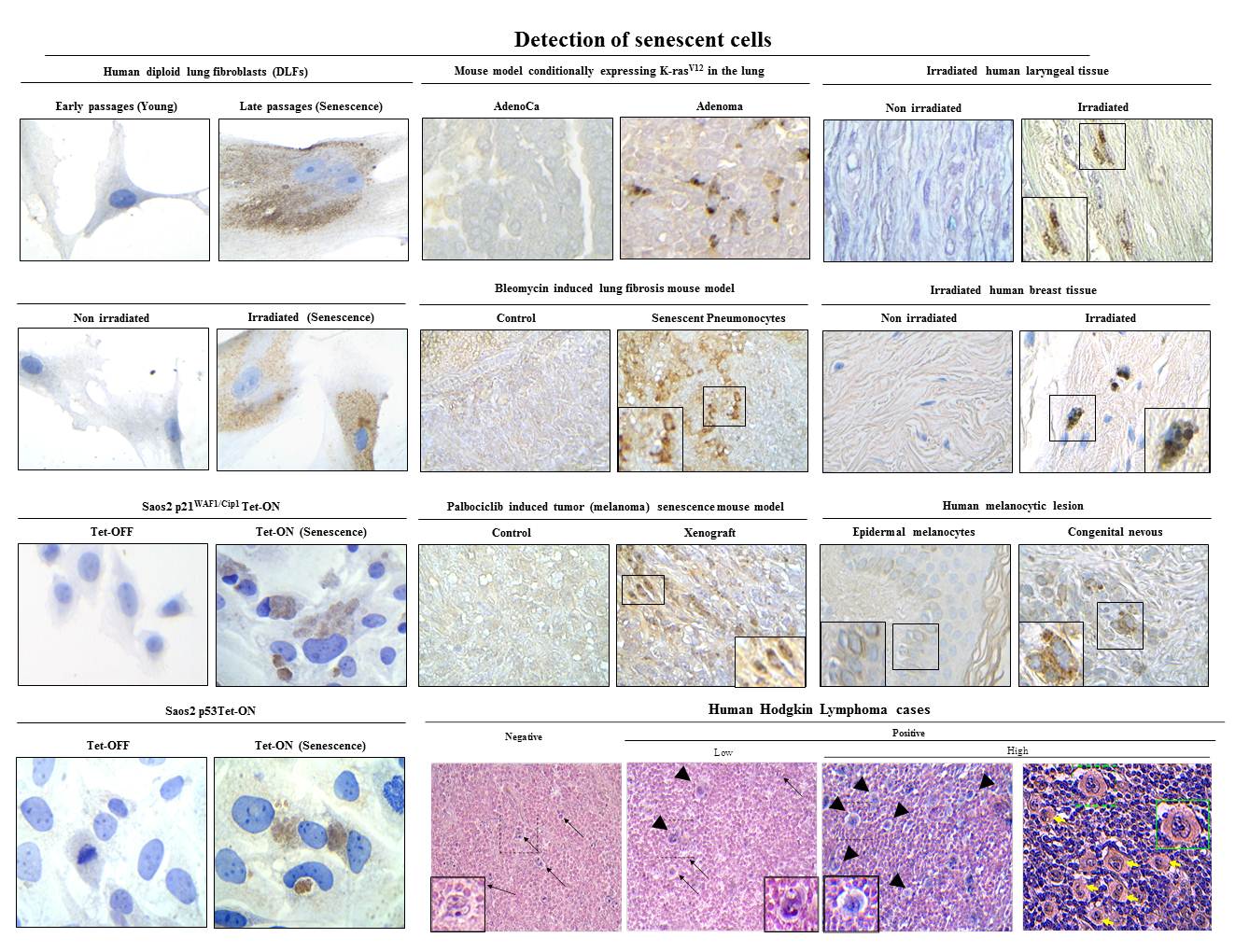
Cellular Senescence: Defining a Path Forward
The Hallmarks of the Senescence Phenotype. Senescent cells exhibit the following four interdependent hallmarks: cell-cycle withdrawal, macromolecular damage, secretory phenotype (SASP), and deregulated metabolism (1).
1) Gorgoulis et al. Cellular Senescence: Defining a Path Forward. Cell 2019, 179(4):813-827.
Integrating the DNA damage and protein stress responses during cancer development and treatment
A model depicting how oncogene induced replication stress aids the progressive formation of certain hallmarks of cancer (early events: steps 1-5), while paving the way for, angiogenesis, evasion from immune surveillance, invasion and metastasis (late events-6). Specifically, oncogenic activation acts as a force that pushes the cell away from its equilibrium point. Activated oncogenes lead to replication stress either directly by deregulating the replication machinery or indirectly via affecting metabolic pathways (1, 2). DNA lesions resulting from oncogene activation stimulate the DNA damage response pathway to promote repair and impose the tumorigenic barriers of apoptosis and senescence. In the event of a perturbed DNA damage response, cells accumulate genomic instability, proteotoxic and mitotic stress (3, 4). Failure to elicit apoptosis or escape from senescence (5) can lead to oncogenic transformation and primary tumor formation. These early events can also pave the way for later events including angiogenesis, evasion from immune surveillance, invasion and metastasis. This is a link that requires further investigation (see text for details). Strike, hourglass: over time, genomic instability shapes the stages for cancer progression. ?: a potential link that requires further investigation [see text in: ref 1 for details and additional references].
1] Gorgoulis et al. Integrating the DNA damage and protein stress responses during cancer development and treatment. J Pathol 2018, 246(1): 12-40.
The non cell-autonomous role of mutant p53 gain-of-function: reprogramming the microenvironment
Colon cancers cells harboring mutations in TP53 release exosomes that contain miR1246 which in turn are received by surrounding macrophages. The latter alter their phenotypic features, leading to secretion of anti-inflammatory cytokines along with factors implicated in epithelial and mesenchymal transition (EMT), which fuel tumorigenesis in this setting (1, 2).
1) Cooks et al. When mutant p53 fires up. Cancer Cell & Microenvironment 2014, 1: e135.
2) Cooks et al. Mutant p53 cancers reprogram macrophages to tumor supporting macrophages via exosomal miR-1246. Nat Commun 2018, 9(1): 771.
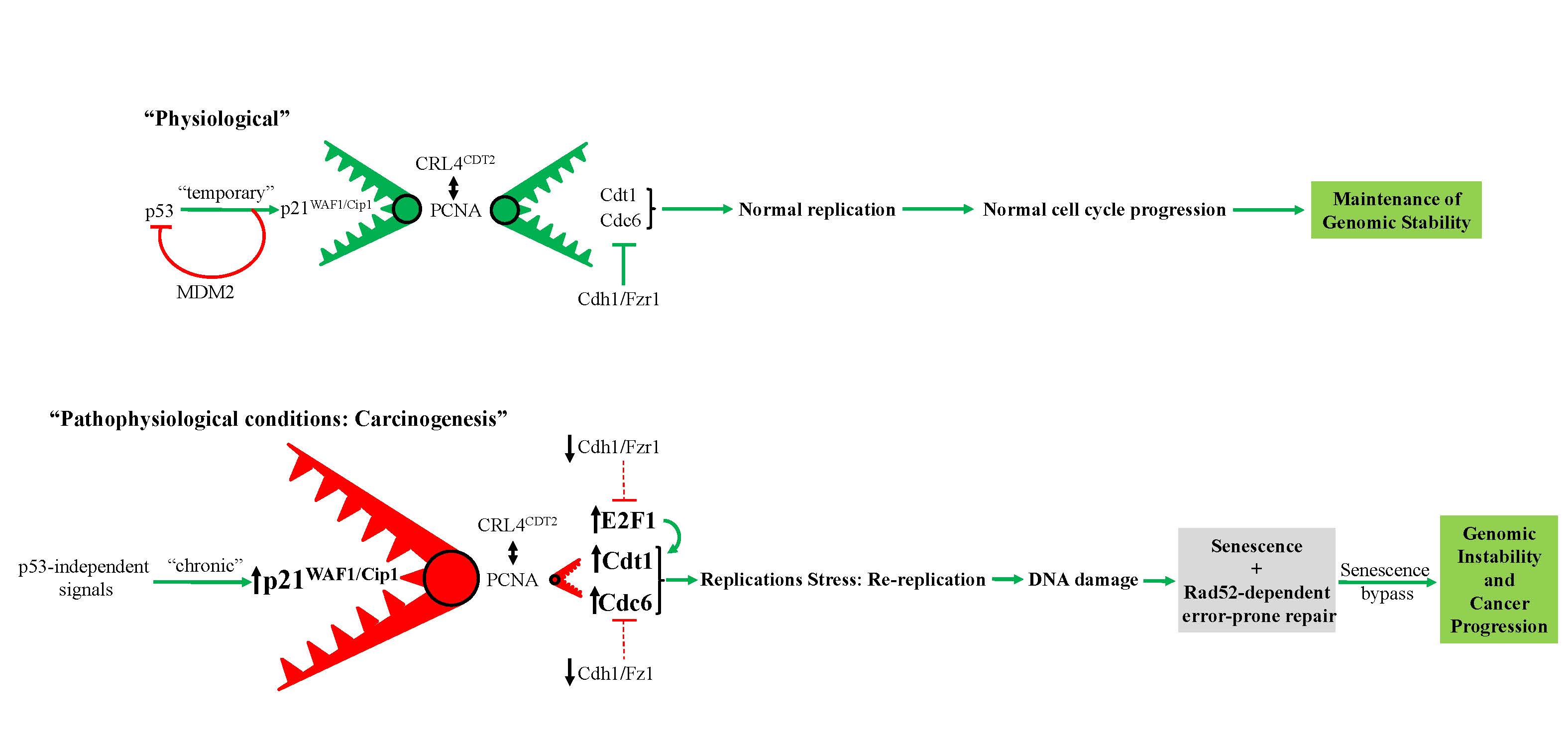
Prolonged expression of p21WAF1/Cip1 in p53-null cells as a driving force for cancer progression
Proposed model depicting prolonged p53-independent p21WAF1/CIP1 oncogenic action. The model shows how p53-independent p21WAF1/CIP1 expression fuels Rad52 dependent error-prone DNA double strand break repair promoting genomic instability (1, 2). Particularly, sustained p21WAF1/CIP1 induction in a p53-independent manner destabilizes the genome through two autonomous but complimentary routes: i) on one hand p21WAF1/CIP1 suppresses its degradation module, CRL4-CDT2, possibly by oversaturating it, as p21WAF1/CIP1 has the strongest PCNA-binding affinity, leaving their other targets, including CDT1, CDC6 and E2F1, free to perform their functions (see below Figure). By upregulating the replication licensing factors CDT1 and CDC6, the cells expressing p21WAF1/CIP1 acquire the capacity to re-replicate, generate DSBs, eventually driving a genome-destabilizing process. (Dashed lines depict ineffective pathway in below Figure), ii) on the hand p21WAF1/CIP1 leads to increased levels of nucleotide lesions mediated by elevated reactive oxygen species (ROS) (2). Given the negative impact exerted by p21WAF1/CIP1 on the error free nucleotide repair mechanisms (BER and NER), a significant proportion of such base lesions escape unrepaired. This creates an additional repair “load” to the error prone repair mechanism of TLS, which is further compromised by p21WAF1/CIP1 overexpression, leading to a decreased SNS load and in favor of DSBs. In turn, this further increases the DSB burden generated also through the re-replication step (i) (1). As components of SDSA are down-regulated, a shift to Rad52-mediated error prone DNA repair takes place by invoking the BIR and SSA repair routes, fueling genomic instability. This repair switch is mediated by a shift in the balance between Rad51 and Rad52 levels as the former is suppressed by E2F4 and the latter is induced by E2F1 (1). Overall these events occur throughout a senescence-like phase during which the error-prone DNA repair process takes place, forming a genetic landscape that allows a subpopulation of p21WAF1/Cip1–expressing cells to escape senescence, generating clones with aggressive features and increased chemo-resistance that promote cancer development. DSB: DNA double strand break, BER: base excision repair, NER: nucleotide excision repair, TLS: translesion DNA synthesis and repair, SDSA: synthesis-dependent strand annealing, BIR: break-induced repair, SSA: single strand annealing.
1) Galanos et al. Chronic p53-independent p21 expression causes genomic instability by deregulating replication licensing. Nat Cell Biol 2016, 18(7): 777-89.
2) Galanos et al. Mutational signatures reveal the role of RAD52 in p53-independent p21-driven genomic instability. Genome Biol 2018, 19(1): 37.
Oncogenic Cdc6 as a molecular switch during cancer development
Model describing the ability of oncogenic Cdc6 to act as a molecular switch. (a and b) Cdc6 acting as a molecular switch at the CDH1 (E-cadherin) (a) and INK4/ARF (b) locus (see Discussion in ref 1). Ori: replication origin.
1) Sideridou et al. Cdc6 expression represses E-cadherin transcription and activates adjacent replication origins. J Cell Biol 2011, 195(7): 1123-40.
|
|
|
Prof. Vassilis G. Gorgoulis
Laboratory of Histology-Embryology
Chair of Clinical Molecular Pathology, Ninewells Hospital and School of Medicine
University of Dundee, Dundee, UK
Biomedical Research Foundation of the Academy of Athens
Faculty Institute for Cancer Sciences, University of Manchester, Manchester Centre for Cellular Metabolism,
EMBO member
European Academy
Academia Europaea member
Intelligencia.ai, 180 Varick Street, 6th Floor, New York, NY 10014, USA
Office Tel: 0030 210-7462352 |









News
Error: No articles to display


